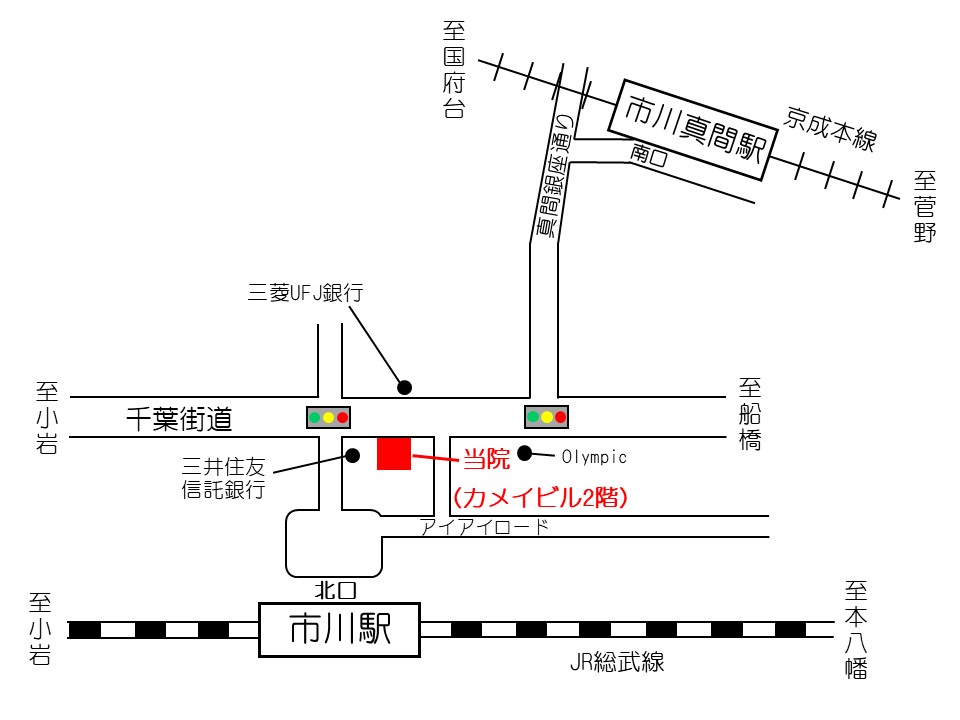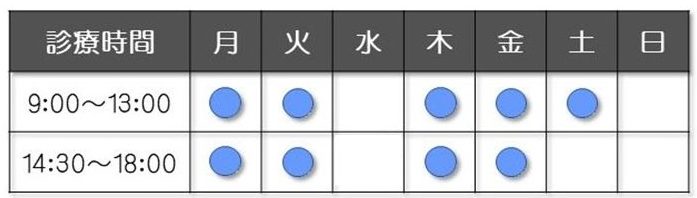

家族性高コレステロール血症
2.原因
3.病型分類
4.診断基準
5.症状と合併症
6.治療方法
家族性高コレステロール血症 (英語:Familial Hypercholesterolemia, FH) は、遺伝的に引き継がれるコレステロール代謝異常症で、悪玉コレステロール(LDL-C)が血中で異常に高くなりやすい疾患です。LDL受容体や関連する遺伝子の変異によりLDL-Cが十分に分解されず血中に蓄積し、動脈硬化を引き起こすリスクが大幅に増加します。このため、FH患者は若年層であっても冠動脈疾患など重篤な心血管疾患を発症する危険性が高く、早期診断および治療が重要です。
FHは主に以下の遺伝子変異によって引き起こされます。
・LDL受容体 (LDLR) 遺伝子
この遺伝子の変異が最も一般的で、LDL受容体の機能不全が原因で、血中のLDL-Cが処理されず高濃度になります。
・アポリポタンパクB (APOB) 遺伝子
LDL粒子が受容体に結合する際に必要なタンパク質です。APOB遺伝子の異常により、LDL受容体との結合が阻害されるため、LDL-Cが蓄積します。
・PCSK9遺伝子
PCSK9はLDL受容体を分解する役割があり、変異により受容体が過剰に分解されることでLDL-Cの増加を引き起こします。
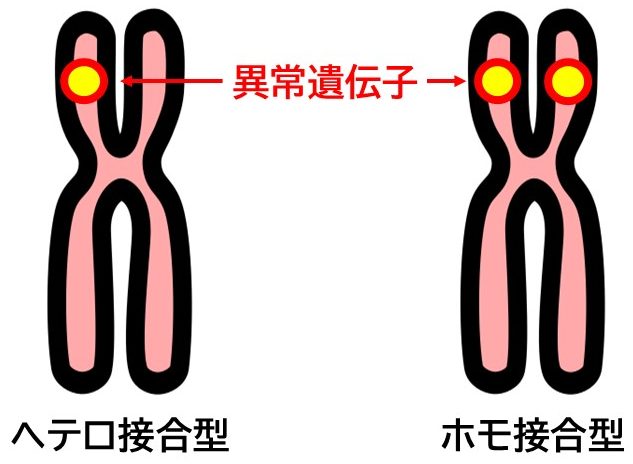
- ヘテロ接合体型FH
片方の親から異常遺伝子を受け継いだタイプで、一般的なFHの形態です。コレステロール値が正常の2倍程度に上昇し、未治療では30代以降に心血管疾患のリスクが高まります。
- ホモ接合体型FH
両親から異常遺伝子を受け継いだまれなタイプで、100万人に1人程度の頻度で認められる指定難病です。コレステロール値が非常に高値となり、極端な高コレステロール血症が生じます。小児期から重篤な動脈硬化が進行し、早期の冠動脈疾患や死亡リスクが顕著に高くなります。
FHの診断は、血中のLDL-C濃度、家族歴、臨床症状、遺伝子検査などをもとに総合的に行われます。日本の診断基準では、次のいずれかが該当する場合に家族性高コレステロール血症と診断されます。
・血液検査によりLDL-Cが成人は180 mg/dL以上、小児は140 mg/dL以上。
・家族にFHの患者や早発性冠動脈疾患がいる。
・腱黄色腫などの特徴的な症状が認められる。
・遺伝子検査でFHに関連する変異が確認される。
FH患者には、腱黄色腫(アキレス腱などにコレステロールが蓄積する病変)や眼瞼黄色腫(眼の周囲に現れるコレステロールの沈着物)が見られることがあります。さらに、若年層で冠動脈疾患、脳卒中、末梢動脈疾患のリスクが非常に高まるため、積極的な治療が不可欠です。
FHの治療は、LDL-Cを目標値まで低下させることを目的に行われます。主な治療は、次のような多面的なアプローチで行われます。
・スタチン
HMG-CoA還元酵素阻害薬であり、LDL-Cを減少させる効果をもつ標準的な治療薬です。
・エゼチミブ
小腸でのコレステロール吸収を抑制する薬剤で、スタチンと併用されることが多いです。
・PCSK9阻害薬
LDL受容体を分解するPCSK9の作用を阻害し、LDL受容体の数を増やすことでLDL-Cを大幅に減少させます。特に重度のFHに有効です。
・リポタンパクアフェレーシス
LDL-Cを除去する血液浄化療法で、ホモ接合体型FH患者や薬剤治療で効果が得られないケースに適応されます。
FHは遺伝性疾患であり、早期に家族内の他のみなさまも検査を受けることが推奨されます。家族全員が早期に診断され、適切な治療を受けることで、心血管リスクを抑制し、寿命を延ばすことが可能となります。
【監修医】

本田 謙次郎(Kenjiro Honda)
市川駅前本田内科クリニック院長/医学博士
東京大学医学部附属病院腎臓・内分泌内科
総合内科専門医・腎臓専門医・透析専門医・厚生労働省認可 臨床研修指導医
略歴
2005 年 東京大学医学部卒、東京大学医学部附属病院・日赤医療センターで初期研修
2007 年 湘南鎌倉総合病院 腎臓内科
2009 年 東京大学大学院医学系研究科(内科学専攻)入学
2013 年 東京大学大学院医学系研究科(内科学専攻)卒業
2014 年 東京大学医学部附属病院 腎臓・内分泌内科 助教
2020 年 市川駅前本田内科クリニック開院・院長就任
その他 宮内庁非常勤侍医、企業産業医等(日本銀行・明治安田生命・日鉄住金建材 ほか)歴任
最新の医学知識をわかりやすく発信し、地域の“かかりつけ医”として健康を支えます。
本記事は一般情報です。診断・治療は必ず医師の診察をお受けください。